Ancient DNA Shows Natural Selection Accelerated During the Bronze Age


David Reich argues that recent human evolution was not dormant after the rise of agriculture but unusually active, especially in and around the Bronze Age. In a discussion of new ancient-DNA work with Ali Akbari, Reich says a large West Eurasian dataset shows widespread directional selection over the past 10,000 to 18,000 years after controlling for migration, drift and admixture. The strongest signals involve immune and metabolic traits, but Reich also reports substantial movement in polygenic scores linked today to cognition, education, pigmentation and body fat, while cautioning that those modern predictors are difficult to interpret in ancient societies.
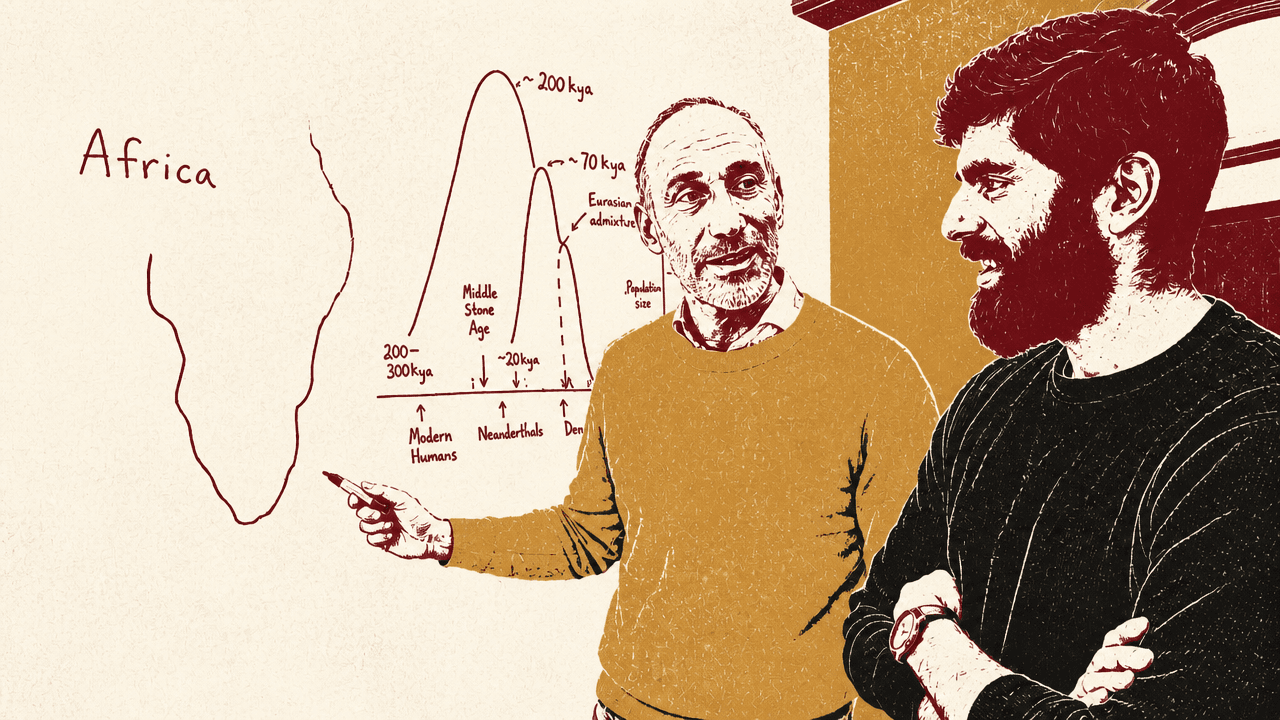
The Bronze Age looks like a biological inflection point, not just a cultural one
David Reich says the long-standing picture in human evolution has been that adaptive directional selection became relatively quiet in recent human history. The ancient-DNA work he discussed, led with Ali Akbari and titled “Ancient DNA reveals pervasive directional selection across West Eurasia,” points in the opposite direction for Europe and the Middle East over the last 10,000 to 18,000 years.
The central finding is not that migrations and population replacement were unimportant. Reich’s group still finds that roughly 98% of observed allele-frequency change is due to other forces, especially genetic drift, migration, admixture, and population structure. But after controlling for those forces, the remaining signal is widespread. In Reich’s phrasing, the genome is “vibrating with natural selection.” Even if selection accounts for only a small fraction of total frequency movement, it appears to be tugging many parts of the genome in systematic directions.
The most surprising timing is the Bronze Age and the period around it. The “cartoon picture,” Reich said, would make the invention and spread of farming the decisive biological shock: hunter-gatherers begin growing plants, living differently, and eating differently. But the genetic data, at least in this West Eurasian dataset, show especially strong intensification later, around 5,000 to 2,000 years ago.
The genetic data, the biological readout, is saying our genome is reacting much more strongly to these events that happened 5,000 years ago.


Reich’s interpretation is that people in this region were “wrenched” into a way of life that differed sharply from hunter-gatherer life, and that the wrenching may have been greater in the Bronze Age than in the initial transition to growing plants. He pointed to higher population densities, closer contact with animals, disease exchange with animals and other humans, more intensive technologies, and urbanizing or proto-urban conditions as plausible pressures. The timing itself is the striking result: biology reacted powerfully not merely to agriculture’s beginning, but to a later transformation in how agricultural and pastoralist societies were organized.
Several of the charts used to explain the result put the strongest movement, inflection, or reversal between roughly 5,000 and 2,000 years before present. The TYK2 tuberculosis-risk chart, for example, showed a derived allele frequency rising from near zero to roughly 0.10 and then dropping sharply toward the present. The intelligence and years-of-schooling polygenic-score charts showed marked upward movement beginning around 4,000 years ago, while a separate strength-of-selection chart for intelligence peaked in the Bronze Age window and showed little signal in the last 2,000 years. Reich described the method behind those charts as dragging a 2,000-year window through the data and re-running the selection analysis inside each window.
That timing shows up across different classes of traits. Immune-related signals are especially enriched among the strongest selection signals. Metabolic traits are also strongly enriched. Some individual examples show sharp changes or reversals in the Bronze Age and Iron Age window. Reich discussed a TYK2 variant associated today with severe tuberculosis risk: in the ancient data it rose to around 9% or 10% frequency in this part of the world and then fell sharply over the last 3,000 years. One possible explanation, which Reich emphasized is speculative, is that the variant may have protected against some earlier pathogen, but became costly once tuberculosis became endemic and sufficiently important.
| Signal or trait | Pattern Reich described | Interpretive status |
|---|---|---|
| TYK2 tuberculosis-risk variant | Rose to roughly 9% or 10%, then fell sharply over the last 3,000 years | Possible pathogen reversal; Reich called the explanation speculative |
| Depigmentation | Strongest movement toward lighter skin roughly 4,000 to 2,000 years ago | Complex-trait signal after ancestry correction |
| Cognitive-performance predictor | Large upward movement over the last 10,000 years, strongest roughly 5,000 to 2,000 years ago | Modern predictor; ancient meaning uncertain |
| Body-fat and type 2 diabetes risk predictors | Directional reduction over the last 10,000 years | Reich connected it to the thrifty-genes idea and food-stability timescales |
He also mentioned variants connected with multiple sclerosis susceptibility, lactase persistence, FADS1/2, ABO blood group, and haemochromatosis. The FADS region had already been known as a strongly selected locus connected with dietary adaptation involving plant and animal fats. The ABO blood system provides a different kind of example: A and B were already present in the ancestor of humans and gibbons, Reich said, yet the B variant increased by up to 10% at the expense of A in the time frame analyzed. Selection here is not always about a new mutation arising from nowhere. Often it is ancient variation moving back and forth under changing conditions.
For skin pigmentation, the signal is a complex-trait result rather than a single-locus story. Europeans became lighter-skinned over the last 10,000 years, and Reich said the strongest period of depigmentation in their data falls roughly between 4,000 and 2,000 years ago, with much less change afterward. That again places the strongest movement after the initial farming transition.
Why population replacement is not the selection signal being measured
The study’s target is narrower than every way genes can become more common. Dwarkesh Patel pressed Reich on the conceptual issue: if one population replaces another, and if genetic differences helped cause that replacement, why not count that as selection? Reich granted that it “could count and may count and probably should count in some respects.” But the paper is trying to identify places in the DNA moving differently from the rest of the genome, not whole-genome ancestry turnover.
A migration can shift the whole genome at once. The steppe migration into Europe around 4,500 years ago is the central example. In parts of Europe, Reich said, 40%, 50%, or even 80% of DNA became related to Yamnaya steppe pastoralists. That produces huge allele-frequency changes across the genome, but those changes do not by themselves identify a particular adaptive variant. The incoming population had different frequencies because it had evolved separately for thousands or tens of thousands of years.
The method therefore looks for a different pattern. Reich described West Eurasian history, for the purposes of this study, as an “archipelago” of small populations in space and time: a pocket in Britain stable for a few hundred years, a pocket in Hungary, a pocket in Italy, each between major migration and mixture events. Within each relatively stable pocket, the question is whether the same mutation tends to move in the same direction. If “all the arrows point in the same direction,” that is evidence for selection.
This is why moments of massive admixture are, statistically, bad moments for detecting natural selection. They produce enormous frequency fluctuations, but the fluctuations are uninformative for distinguishing an adaptive locus from ancestry change. The cleanest test comes when ancestry is relatively stable for long enough to observe smaller, systematic allele-frequency shifts.
The paper’s main methodological move is to control for relatedness and ancestry structure so that these global shifts do not masquerade as local adaptation. Patel summarized the logic as a model with two parts: a genetic relatedness matrix that captures similarity among genomes, bottlenecks, drift, and admixture; and an additional term asking whether a specific location in the genome is better explained by a selection coefficient over time. Reich said that was “precisely right.”
The dataset made this possible in a way earlier ancient-DNA work did not. Ancient DNA had already transformed population history, Reich said, because one genome contains information about many ancestors: parents, grandparents, great-grandparents, and so on. That makes even a single genome powerful for placing a person in a population-history tree. But selection on a particular variant is different. One person supplies only one or two copies of a variant. To estimate frequency changes with resolution, very large numbers of ancient individuals are needed.
The scale of available ancient genomes has changed the question. A chart attributed to the Allen Ancient DNA Resource tracked published ancient human genomes with genome-wide data from 2010 to 2024, rising exponentially to 10,067, and noted that the latest release contained 17,634 published ancient genomes. Reich said his group’s study reports new data from about 10,000 individuals and analyzes a total dataset of about 16,000 ancient individuals distributed over the last 18,000 years. Including modern people, he described a dataset of about 22,000 individuals.
That dataset is geographically narrow by design and by necessity. It covers Europe and the Middle East, which Reich said are not more important than other parts of the world but have, for historical reasons, produced perhaps 70% or 80% of the ancient-DNA literature so far. That makes the region an unusually good natural laboratory for asking how genomes responded over time as environments and lifeways changed.
The strongest signals are immune and metabolic, but behavior is not absent
The most confidently detected selection signals are not evenly distributed across kinds of traits. David Reich said the group looked at roughly a hundred traits with genome-wide association studies, including immunity, autoimmunity, behavior, metabolism, and related categories. Among the strongest selection signals, immune traits were enriched by roughly four- to five-fold. Metabolic traits, including variants connected with obesity, fat traits, and type 2 diabetes, were also strongly enriched. Behavioral and psychiatric traits did not show comparable enrichment among the strongest single-position signals.
That is not the same as evidence that behavior was not under selection. Reich’s explanation depends on genetic architecture. Immune traits are often affected by smaller numbers of loci with larger effects. Behavioral traits are influenced by very large numbers of variants of weak effect. A scan that is most powerful for large single-locus signals will naturally detect immune-related changes more readily than diffuse behavioral changes.
The data, Reich said, provide “clear evidence” of selection on behavioral traits too, but the signals are weaker at individual sites. The relevant test is polygenic: aggregate many variants associated today with a trait, weight them by their effects, and ask whether the resulting score moved directionally through time after correcting for ancestry.
The cognitive-performance result is the most sensitive of the complex-trait findings, and it carries two caveats that matter throughout. First, the ancient-DNA analysis is about West Eurasia, not all humans everywhere. Second, the predictors come from modern genome-wide association studies and are context-dependent; a score that predicts intelligence-test performance, years of schooling, or household income today is not literally measuring ancient intelligence tests, ancient schooling, or ancient income.
Reich described using genetic variants associated today with performance on intelligence tests in white British people, and also closely correlated predictors for years of schooling and household wealth. He emphasized that these are “crazy traits in the past” in the literal sense that there were no intelligence tests, no modern school, and no household wealth as measured today. The combinations of genetic variants that predict those outcomes today nevertheless moved systematically in ancient populations.
The movement Reich described is large: about one standard deviation on the scale of modern variation over the last 10,000 years. Patel translated that into percentile terms: one standard deviation above the median is roughly the 85th percentile, so a one-standard-deviation shift in a polygenic score is a major effect. Reich accepted the magnitude and added that migration effects are even larger in some plotted trajectories.
For example, in the intelligence polygenic-score chart, he said European hunter-gatherers are estimated around three standard deviations below the modern mean, early farmers around the mean, and steppe pastoralists lower again. Those jumps are not the selection result; they largely reflect migration and different population ancestries. The selection test asks whether, in addition to those jumps, there is a consistent directional push within relatively stable population pockets.
| Polygenic score | Movement Reich emphasized | Caution Reich attached |
|---|---|---|
| Intelligence-test performance predictor | About a standard-deviation-scale movement over 10,000 years; strongest selection signal in the Bronze Age window | Measured in modern people; ancient populations had no tests |
| Years of schooling predictor | Similar direction to the intelligence-related predictor | There was no modern schooling in the ancient setting |
| Household income predictor | Moved in the same broad direction | Modern wealth is not an ancient trait |
| Body fat / BMI / type 2 diabetes risk predictors | Reduced over the last 10,000 years | Interpretation depends on food access and timescale |
For the intelligence-related polygenic score, the strongest selection occurred between roughly 5,000 and 2,000 years ago, especially 4,000 to 2,000 years ago. In the last 2,000 years, Reich said, there is essentially no evidence of natural selection on that score. He found this counterintuitive: if one came in expecting any selection on such a trait, one might guess it would be strongest during the last two millennia, with industrialization or increasingly complex societies. Their data instead place the strongest signal in the Bronze Age and adjacent period.
The years-of-schooling score raised a separate concern: maybe this is an artifact of European GWAS rather than a real biological signal. Reich said they tested this by using a study of educational attainment in Chinese people in China. They asked whether variants that predict more years of schooling in China today correlate with the trajectory of those same variants in ancient Europeans. These populations were, for the purposes of the test, essentially disconnected over the relevant timescale. Yet Reich said they found a five- or six-standard-deviation correlation, about as strong as when using European effect sizes. That convinced them the signal was not simply a European GWAS artifact. In Patel’s summary, the same parts of the genome seem to robustly predict “the kind of thing” that leads to more years of schooling in people today.
The cognitive signal may not be intelligence in the simple sense
David Reich repeatedly cautioned against reading the cognitive-performance and educational-attainment results too literally. The genetic predictor of years of schooling is correlated with many traits that are not school or intelligence: age at first childbirth in women, obesity, walking pace, household wealth, and others. He suggested that the selected dimension might be something more general, perhaps executive function, delayed gratification, planning, or another correlated trait that manifests differently across societies.
He brought up a 2017 Icelandic study by Kong and colleagues, “Selection against variants in the genome associated with educational attainment.” The visible abstract said an educational-attainment polygenic score was associated with delayed reproduction and fewer children overall, with a stronger effect in women, and that the average score had been declining by about 0.010 standard units per decade among Icelanders born between 1910 and 1990. Reich described this as about a 0.1-standard-deviation decrease over a century, a large effect over such a short evolutionary timescale. He corrected himself to specify that the study was about selection against genetic predictors of years of schooling, not necessarily intelligence itself.
One possible interpretation is that the score partly measures a reproductive strategy trade-off: having children earlier and more of them, with lower investment per child, versus delaying reproduction, accumulating more resources, and investing more in fewer children. In a “time of plenty,” he suggested, the high-fertility strategy might win even if the children individually receive less investment. That would produce selection against variants associated today with more education, without implying a straightforward selection against intelligence.
Dwarkesh Patel raised the “collective intelligence” hypothesis: as societies become more specialized, each individual might need to know less, so selection could turn against individual cognitive ability. He also noted the intuitive counterpoint from Joseph Henrich’s work: hunter-gatherers must know enormous amounts about food processing, shelter, fire, terrain, and survival; modern specialists may need a narrower knowledge base. Reich said the data go against some expectations. If one had asked Henrich beforehand, Reich guessed, Henrich might not have made a strong prediction but might have expected hunter-gatherers to score high on the relevant predictor. The ancient-DNA result instead shows a strong later increase in the genetic variants associated with modern measures.
Reich also questioned whether societies in the past valued intelligence in the way modern societies do. He pointed to the Hebrew and Christian Bible, Homer, and other ancient texts as emphasizing strength, courage, beauty, religiosity, or other virtues rather than “smarts” in the modern educational sense. Patel objected that the Old Testament was being written during precisely the period when the study finds selection on the intelligence-related score was high. Reich accepted the timing but maintained that explicit cultural valuation of IQ-like traits was not obviously central.
The broader point is that a trait can be useful in one ecological or social configuration and less useful, or costly, in another. Reich extended the logic to psychiatric traits. Schizophrenia and bipolar risk seem obviously bad if one looks only at severe disease. But subclinical traits connected with anxiety, imagination, visions, creativity, or unusual cognition may have been valued or useful in some religious or shamanistic contexts. He mentioned Julian Jaynes’s theory of the bicameral mind only in response to Patel’s prompt, and did not endorse it as a settled explanation. His point was narrower: variants contributing to psychiatric risk may sit on broader spectra where different points on the spectrum have different payoffs under different cultural systems.
Selection against body fat fits a food-stability story, but only at the right timescale
For body fat and metabolic disease risk, David Reich described a clear directional trend over the last 10,000 years in Europe and the Middle East: selection reduced combinations of variants associated today with obesity, body mass index, fat mass, waist-to-hip ratio, and type 2 diabetes risk. He put the magnitude at roughly one standard deviation on the scale of modern variation.
The candidate explanation is the “thrifty genes” hypothesis. In hunter-gatherer settings, storing fat may help survive boom-bust food access. In farming environments with more stable food stores, those same tendencies may become less advantageous, producing selection against higher body fat. Reich noted that Europeans are relatively more genetically protected against type 2 diabetes than some other populations, such as African-Americans and Native Americans, and suggested this may reflect longer exposure to agriculture and more stable food availability.
Dwarkesh Patel pointed out that this cuts against a familiar story: hunter-gatherers are often said to have more stable diets because their food sources are diverse, they are not dependent on a single cereal crop, and they can move when resources shift. Agricultural societies, by contrast, can suffer famines when crops fail. If selection moved against fat storage after agriculture, Patel said, that suggests agricultural life was at least more stable in the relevant sense than hunter-gatherer life.
Reich’s answer turned on timescale. In some hunting societies, he said, people may gorge after a successful hunt and then go days without meat. Body fat helps bridge short boom-bust cycles. Agricultural famine may be more common over multi-year intervals, and bones from some farming communities show stress, perhaps from famines every three or five years. But fat accumulated after a hunt does not carry someone through a famine three years later. Selection on fat storage may respond to short-term nutritional volatility rather than long-cycle famine risk.
This is a recurring feature of Reich’s reasoning: whether selection sees a pressure depends not only on the pressure’s severity but on its timing, recurrence, and relation to reproduction and survival. The same society can have more catastrophic famine risk but less day-to-day or week-to-week nutritional volatility.
Population size is probably not why selection accelerated
Dwarkesh Patel asked whether the Bronze Age might have mattered because human population size became large enough for more variants to be “visible” to selection. David Reich said this is unlikely. Population size matters for selection in small populations because allele frequencies randomly “bop around” from generation to generation through drift. In a population of 1,000, frequency fluctuations are on the order of one over 1,000 per generation; a selection coefficient smaller than that can be drowned out. But the selection coefficients Reich’s study detects are often on the order of half a percent or 1%, which are strong enough to operate in populations of 1,000 or 10,000.
Once populations are on the order of a million, Reich said, essentially every mutation that can occur will occur within a few generations. In the present world of 8 billion people, with perhaps 30 new mutations per person per generation, every possible point mutation occurs many times each generation. But for the strong selection coefficients observed in this study, mutation supply and population size are not the limiting factor. Time is.
Patel summarized the point as: once a population passes a threshold size, the dominant factor is timespan rather than population size. Reich answered, “Correct,” and added that the point is “not widely understood.”
This matters for interpreting the Bronze Age. If the relevant selection coefficients are strong, they could have operated in earlier, smaller populations too. The acceleration therefore points less to population size alone and more to environmental and social change: new disease ecologies, new diets, new densities, new reproductive regimes, or other selection pressures.
Reich also generalized the point to complex traits. Human populations contain large amounts of standing variation. If one set all height-increasing variants in the same direction, he said, the result would be absurdly tall, “as tall as a tall building.” The same applies to schizophrenia risk, obesity risk, and other polygenic traits. For complex traits governed by many variants, the raw material for movement often already exists. A population pushed into a new environment can shift its mean over hundreds or thousands of years by changing frequencies among existing variants.
Some traits are different. Lactase persistence or sickle-cell protection can depend on single important mutations that may not yet exist in a small population. In a population of 10,000, one may have to wait dozens or hundreds of generations for the right mutation to arise. But for the broad polygenic traits at the center of the paper, Reich’s emphasis is on standing variation and time under selection.
No single genetic switch explains behavioral modernity
Dwarkesh Patel connected the recent-selection results to an older puzzle: if modern humans underwent a “cognitive revolution” around 50,000 to 100,000 years ago, with representational art, bead necklaces, cave drawings, and increasingly rapid tool innovation, should there be a genetic sweep that distinguishes modern humans from archaic humans?
David Reich said his 2016 work with Swapan Mallick and colleagues looked hard for such sweeps: places in the genome where all or nearly all people living today share a recent common ancestor, perhaps 100,000 or 200,000 years ago, indicating a key mutation that rose to high frequency. They found nothing more recent than roughly 400,000 or 500,000 years ago. Reich called that “a crazy result.” There does not seem to be a single key biological change that swept through all modern humans in the period when much of the material-culture evidence of behavioral modernity appears.
That does not mean no biological adaptation occurred. Reich’s alternative is that any adaptation may have been polygenic: many mutations shifting together, with no single mutation rising to fixation. The recent-selection paper strengthens that way of thinking. It shows that important trait shifts can be distributed across many loci and can be missed by methods designed to find classic sweeps.
The absence of fixed differences also bears on modern human diversity. Reich said the relevant population 100,000 to 50,000 years ago is ancestral to West Africans, most East Africans, and all non-Africans, though some African populations such as Khoisan groups and Central African rainforest hunter-gatherers have substantial ancestry from lineages that diverged perhaps 200,000 years ago. But all these groups today are capable of “going to college” and doing what everyone else does. Reich’s point was that there is no evidence of a key mutation present in some groups and lacking in others that explains modern cognitive capacity.
That leads to the “long fuse” problem. If the genetic ingredients for farming, state-building, and symbolic culture were in place tens of thousands of years earlier, why did agriculture appear only after the Ice Age? Reich said he asks climate scientists and archaeologists this often. Their answer, as he reported it, is that the Holocene was not merely warm but unusually stable on a two-million-year timescale. Isotopic signatures from pond bottoms and other climate records suggest much lower fluctuation year to year, decade to decade, and century to century. Once that stability arrives around 12,000 years ago, agriculture appears independently or semi-independently in multiple places.
Patel found the climate explanation surprising because farming arose in very different environments: maize in the Americas, cereals in the Old World, and other systems elsewhere. Reich agreed that it is surprising. But he argued the fact remains: descendants of a common ancestral population dispersed to West Africa, East Africa, the Americas, Europe, South Asia, East Asia, New Guinea, and elsewhere, and only long after dispersal did agriculture ignite in multiple places. That implies the cognitive and behavioral toolkit was already there, waiting on ecological or climatic conditions rather than a new human-specific genetic switch.
Patel asked whether earlier farming civilizations might have existed and disappeared without leaving a record, perhaps lacking metallurgy or other features that later allowed population explosion. Reich doubted this. He said archaeology would likely show it, and pointed to the Americas as proof that metallurgy was not required for monumental societies. Teotihuacan, he said, is “totally as impressive as ancient Egypt,” despite being built without metal in the Old World sense. Patel added that it was also without animals and wheels. Reich’s point was narrower than a general theory of all societies: impressive, large-scale material remains can exist without Old World metal, draft animals, or wheels, so a large earlier agricultural civilization would be hard to hide completely.
The Neanderthal problem has three layers: consensus, discomfort, and conjecture
The standard genetic model, as David Reich described it, is straightforward at the whole-genome level. Neanderthals and Denisovans descend from a common ancestral population around 500,000 to 600,000 years ago, and that lineage separated from the ancestors of modern humans perhaps 700,000 to 800,000 years ago. Whole-genome data strongly support Neanderthals and Denisovans as sister groups.
Reich’s discomfort begins with the facts that do not sit easily with a simple version of that tree. Neanderthals and modern humans share Middle Stone Age or Middle Paleolithic stone-tool technologies, especially Levallois technology, which he described as a cognitively distinctive way of making stone tools from carefully prepared cores. This tradition is shared in Africa and Europe but absent in East and South Asia in the same way. Neanderthals also have mitochondrial DNA and Y chromosomes that cluster with modern humans rather than Denisovans. Existing work explains this through interbreeding from a lineage related to modern humans into Neanderthals around 200,000 to 300,000 years ago, contributing perhaps 5% of Neanderthal DNA.
That 5% explanation is accepted in the field, but Reich finds it odd as a full story. If only 5% of Neanderthal ancestry came from this modern-related source, it is surprising that both the mitochondrial DNA and the Y chromosome would jump to 100% frequency. He described the chance intuition as roughly 5% times 5%, a low probability, while acknowledging that this is what the field has largely come to believe because the evidence accreted in that direction.
The third layer is Reich’s provisional model. After the main recording, he sketched it on a whiteboard: first the standard tree of Denisovans, Neanderthals, and modern humans; then a 200,000- to 300,000-year-old gene-flow arrow from the modern-human lineage into Neanderthals, labeled around 5%; then a geographic model in which a population associated with Levallois or Middle Stone Age technology expands into both Europe and Africa.
| Layer | Claim in Reich’s account | Status |
|---|---|---|
| Consensus whole-genome tree | Neanderthals and Denisovans are sister groups, split from modern humans earlier | Standard genetic model |
| Source of Reich’s discomfort | Neanderthals share modern-like mitochondrial DNA, Y chromosomes, and Middle Stone Age / Levallois technology | Accepted observations, hard to fit intuitively with only a minor gene-flow event |
| Provisional model | A modern-related expanding population may have carried technology and uniparental markers while being genetically swamped by local archaic groups | Speculative; Reich said it is an idea he is playing with and may be wrong |
Reich did not present the map as a settled location of origin. On the whiteboard, he described a possible population associated with the Middle Stone Age or Levallois transition and mentioned places such as the Caucasus, Georgia today, East Africa, the Middle East, or Northeast Africa as part of a speculative reconstruction of where such a process might have begun or moved through. The geography was tentative; the model’s point was not a pinned homeland but a possible expansion carrying technology, some ancestry, and perhaps uniparental markers into landscapes already occupied by archaic humans.
The proposed population expands in multiple directions. In Europe, it encounters local archaic humans. As it spreads, it mixes with them and becomes genetically swamped by local archaic ancestry, ultimately producing Neanderthals: mostly archaic in whole-genome ancestry, but carrying a modern-related cultural tradition, mitochondrial DNA, Y chromosome, and perhaps some genetic adaptations. In Africa, the same expanding population mixes with more divergent archaic African groups and contributes to the formation of anatomically modern humans.
Reich invoked a process known from simulations and studies of other species: when one population expands into a landscape occupied by another, even small amounts of interbreeding at the wavefront can lead to massive introgression of local genes. Pioneers repeatedly mate with locals; because locals are numerous, the expanding group’s genome can be swamped even as its culture or social system keeps moving. By the time the wave reaches the far side of the landscape, it may be mostly local genetically.
To make the idea intuitive, Reich compared it with Yamnaya ancestry and Indo-European languages in South Asia. As Yamnaya-related groups expanded, mixed, and diluted, the ancestry component in many Indian groups became small: sometimes 20%, 10%, 5%, or less. But it can still serve as a “tracer dye” for language and cultural transmission. A 5% genetic contribution, in this view, should not be dismissed as unimportant. It may trace the population that brought a major cultural package.
The Neanderthal version is more radical: Neanderthals might be thought of as, in some sense, culturally modern humans who became genetically swamped by European archaic humans. Reich did not present this as established. But he said it could explain why Neanderthals share Levallois technology, mitochondrial DNA, and Y chromosomes with modern humans while still clustering genome-wide with Denisovans.
A key piece in the puzzle is Sima de los Huesos in Spain, dated to roughly 300,000 to 400,000 years ago. Reich said those individuals have nuclear genomes that look Neanderthal-like, but mitochondrial DNA and Y chromosomes that are Denisovan-like. That pattern makes it look as if a population related to modern humans later pushed into a Sima-like population, displaced its mitochondrial DNA and Y chromosome, but left much of the nuclear genome intact.
The African side of the model relies not on ancient African DNA, which is largely unavailable at those depths, but on analyses of modern DNA. Reich said multiple studies of modern African and non-African genomes find that the ancestors of anatomically modern humans were not a homogeneous population. Instead, the data look like a split more than a million years ago into at least two, and probably more, groups, followed by major admixture a few hundred thousand years ago. In his whiteboard model, an “early modern” lineage might contribute around 80%, while an archaic African lineage contributes around 20%, though he emphasized different papers fit different models and the geography is unknown.
The larger implication, if the model were right, is that the formative event for both Neanderthals and modern humans may be older than the symbolic explosion often placed around 50,000 to 100,000 years ago. The Middle Stone Age or Levallois transition around 300,000 to 400,000 years ago may be equally important. If so, Neanderthals are not merely a separate archaic lineage with a small amount of modern-related ancestry. They may share in the same cultural and perhaps biological expansion that also helped form modern humans.
The method worked because the field finally had enough data to stop seeing only the largest effects
David Reich framed the new paper as the fulfillment of an old promise of ancient DNA. The field had succeeded spectacularly at reconstructing population history: migrations, unexpected ancestry shifts, mixture, sex-biased processes, and replacements not predicted by archaeology alone. It had not delivered as much on biology, because tracking a particular variant’s frequency requires many ancient individuals.
Earlier selection scans seemed to confirm the pessimistic view. In 2015, Reich and colleagues including Iain Mathieson analyzed about 200 ancient Europeans and Middle Easterners and identified 12 positions they were convinced had changed too much over time to be explained by chance. That was exciting, but the hope was that larger datasets would produce many more discoveries. A 2024 Copenhagen-led study, described by its title as “The selection landscape and genetic legacy of ancient Eurasians,” analyzed more than 1,600 imputed ancient genomes and found 21 highly differentiated positions. Reich said that was nearly twice the 2015 number but still disappointing given the increase in data. It suggested the field might be approaching an asymptote, perhaps because adaptive directional selection really had been quiet.
Akbari’s study changed both scale and method. Reich said their analysis considered about 10 million variable positions in roughly 22,000 people, including around 16,000 ancient individuals. They asked whether a selection model improved prediction of genotypes beyond what could be predicted from each person’s relatedness to all others in the dataset. The simplifying assumption was constant selection over time and geography. Reich called that assumption “dumb” because true selection changes through time, but it gave them a simple first test: does adding a directional-selection term explain the data better than relatedness alone?
The reported counts are not a single harmonized number. In one exchange, Reich corrected Patel and said the study had about 7,200 positions where they were 50% confident, implying about 3,600 real signals without knowing which individual positions were real. In the methodological discussion, he described a separate counting exercise meant to avoid double-counting densely packed, interfering nearby signals: after counting only one in each place and blanking out others, he said they found at least 479 independently pushing positions at 99% confidence and about 3,800 independent positions by a criterion of more than 50% confidence. The important point is not that all these figures are interchangeable, but that the scan moved from dozens of discoveries in earlier work to hundreds or thousands of candidate signals, depending on the confidence threshold and independence criterion.
| Count Reich gave | Threshold or definition in the source | How to read it |
|---|---|---|
| 7,200 positions | 50% confidence in one exchange with Patel | Candidate positions; Reich said roughly half would be real, without knowing which half |
| 3,800 positions | More than 50% confidence by a later independent-position counting criterion | A separate count after addressing dense local clustering and interference among nearby signals |
| 479 positions | 99% confidence by the independent-position criterion | The most stringent set Reich described |
The validation step was crucial. Reich said they tried for years to make the results disappear, assuming at first that something must be wrong. The independent check came from genome-wide association studies. In the UK Biobank, about 15% of 10 million positions are predictive of at least one measured trait. If the ancient-DNA selection statistic is meaningful, positions with stronger selection signals should be enriched for trait-associated variants. That is what they found. As the selection statistic rose, enrichment for trait-associated variants rose too, reaching about a five-fold enrichment. Above a statistic around 5, Reich said, 60% or 70% of mutations affected a trait, compared with 15% at random, and enrichment plateaued.
This gave them a calibration. If a selection-statistic bin is halfway to the plateau, Reich said, roughly half the mutations in it are real selection signals. If it is three-quarters of the way, about three-quarters are real. If it is 99% of the way, about 99% are real. Akbari’s idea, as Reich described it, abandoned the traditional approach of assigning significance solely from the scan itself and instead used independent trait association as a way to read off the probability that signals were real.
They were also concerned about a confound: background selection. Genes are both where disease-associated variants tend to occur and where selection against newly arising deleterious mutations is concentrated. That shared structure could create a false enrichment. Reich said they repeated the enrichment analysis within slices of the genome equally affected by background selection and got the same pattern. They also repeated it within frequency-matched mutation sets, since power varies by allele frequency, and again saw the same plateau.
The technical foundation was years of laboratory scaling. Sequencing costs fell enormously: Reich said roughly a million-fold since the late 2000s and another one to two orders of magnitude from 2010 to today. Ancient samples often contain less than 10%, sometimes less than 1%, human DNA; the rest is mostly microbial DNA from bacteria and fungi that colonized the body after death. In-solution enrichment made it economically feasible to target useful human positions. Reich described washing ancient DNA over artificially synthesized short DNA fragments targeting more than a million informative positions, many chosen for biological interest. The target fragments bind the relevant ancient DNA, enriching the sequencing output for useful human variation rather than microbial background.
Roboticization and industrialization then pushed throughput from tens of samples per year to hundreds, thousands, and in Reich’s lab more than 5,000 genome-scale ancient individuals per year. The field went from around 10 ancient human genome sequences in 2010 to more than 20,000 reported sequences by the time of the discussion. Reich’s conclusion was simple: the questions one could ask in 2014 are not the questions one can ask now.